Obhajoba bakalářské práce - Kamila Herková
Created by Kamila Klavíková
🔹 17. 9. 2019
🔹 Porovnámí dostupných algoritmů predikce sestřihu rostlinné mRNA
🔹 obor bioinformatika
#PřF UK, #bioinformatika, #biologie, #prezentace

Porovnání dostupných algoritmů predikce sestřihu rostlinné mRNA
Bakalářská práce
Kamila Herková
vedoucí: doc. RNDr. Fatima Cvrčková, Dr.
Sestřih
- Syntéze proteinů předchází přepis DNA do prekurzorové mRNA (pre-mRNA), která není plně funkční.
- Proto dojde k úpravě zvané sestřih, kdy z pre-mRNA vznikne zralá mRNA.
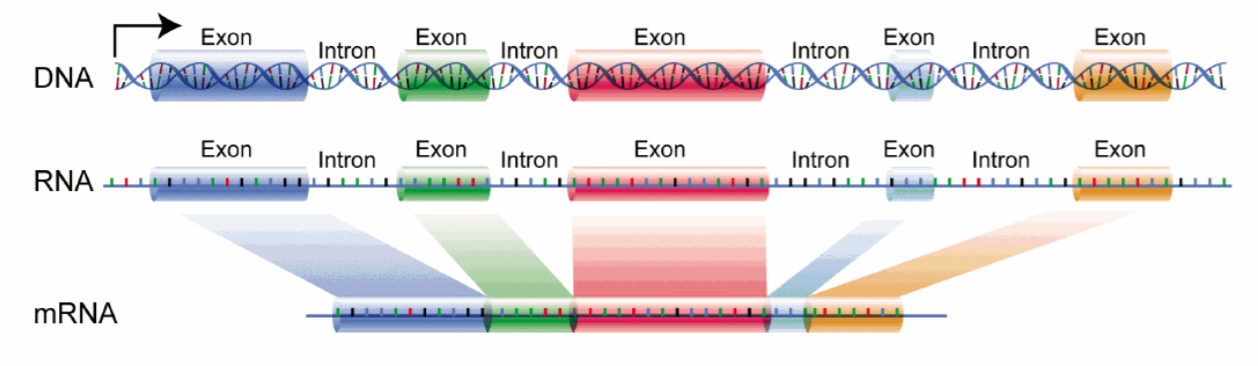
Alternativní sestřih
- Z jedné pre-mRNA může vzniknout několik transkriptů s různými sekvencemi.
- Přispívá k rozmanitosti bílkovin, neboť jich je několikanásobně více než genů.
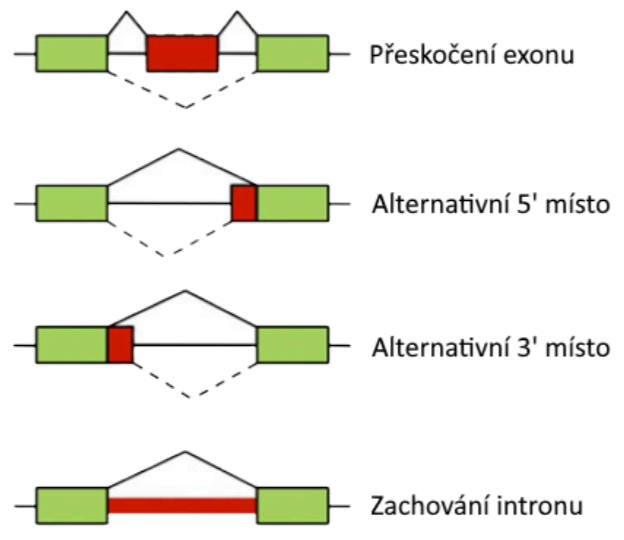
Metody pro predikci genů
- Hledají v zadané sekvenci úseky kódující geny: exony, introny a další funkční elementy, například nepřekládané oblasti.
- Využívají obvykle skryté Markovské modely.
Metody založené na sestavení transkriptomu
Tyto algoritmy jsou jedním z kroků procesu analýzy RNA dat (Song et al., 2018). K sestavení transkriptomu jsou dva hlavní přístupy.
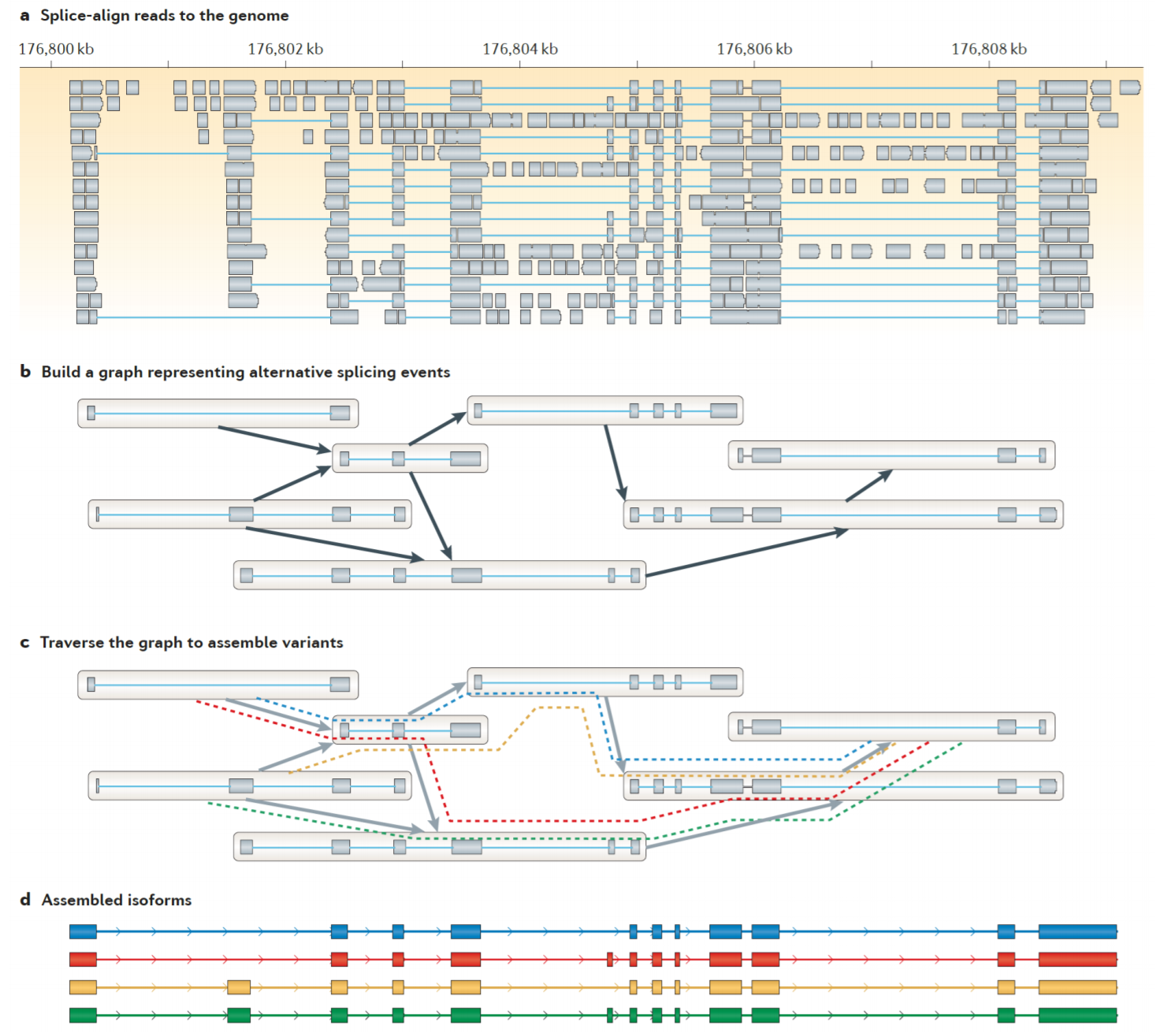
Predikce s referenčním genomem
Hlavní kroky algoritmů: - Nejprve zarovnají čtení na zadaný genom. Například pomocí nástroje TopHat.
- Následně budují graf překryvů zachycující všechny možné izoformy.
- Na závěr prohledají graf a sestavují jednotlivé izoformy.
|  |
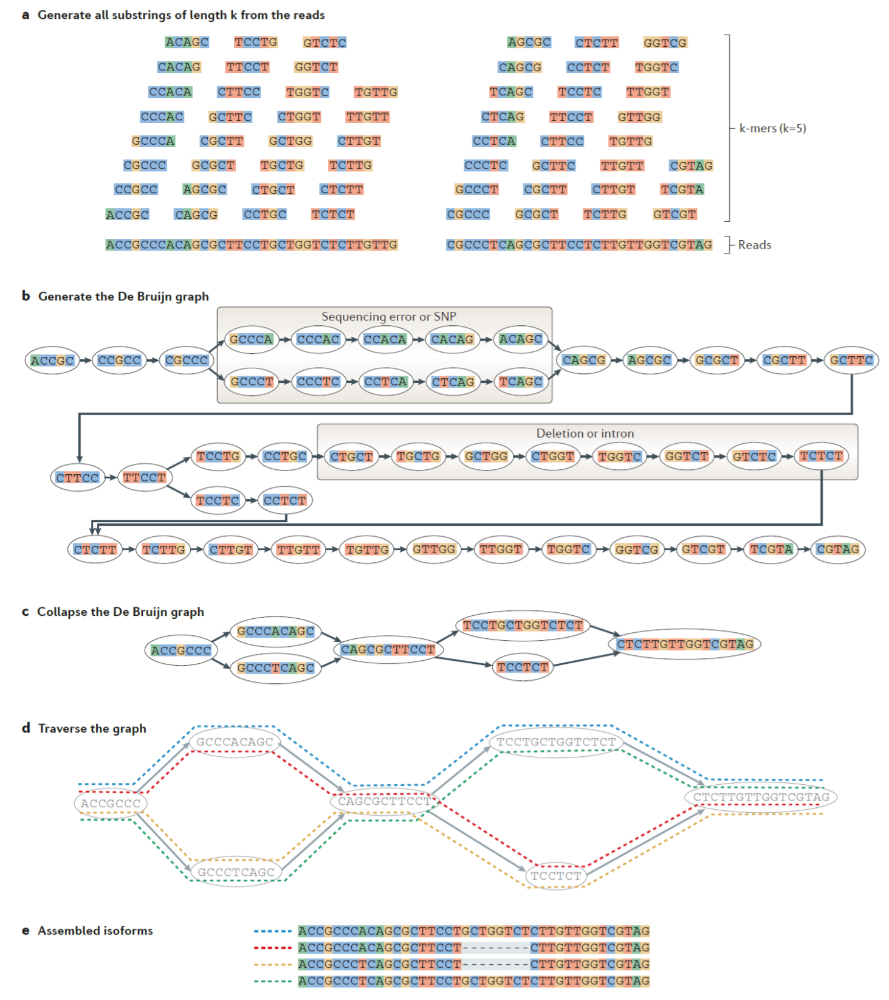
Metody de novo
Používají se v situaci, kdy nemáme dostupný kvalitně sestavený genom. Například při analýze nemodelového organismu s ekologickým nebo evolučním významem (Haas et al., 2013). - Vygenerují všechna podslova délky k z jednotlivých čtení.
- Z nich sestrojí de Bruijnův graf a provedou zjednodušení.
- Prohledáním grafu sestaví jednotlivé transkripty.
|  |
Markovské modely
- Systém tvořený stavy x1, ..., xn, stav se mění v krocích. Nový stav závisí pouze na předchozím stavu, nezávisí na historii. Je popsán stochastickou maticí.
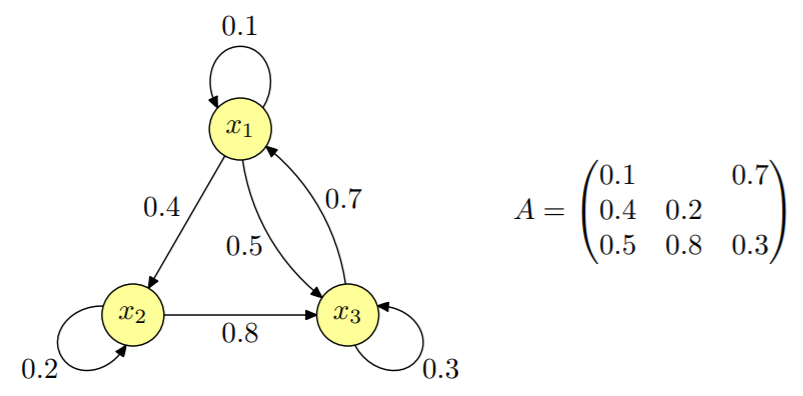
- Vývoj pravděpodobnostní distribuce u dostaneme jako u, Au, A2u, A3u, ..., tedy opakovaným násobením maticí A.
- Lze analyzovat metodami lineární algebry (vlastní čísla, atd.).
Neuronové sítě
Skryté Markovské modely
- Dodatečná informace ve formě pozorování. Známe pravděpodobnosti, že nastaly jednotlivé stavy vzhledem k pozorováním.
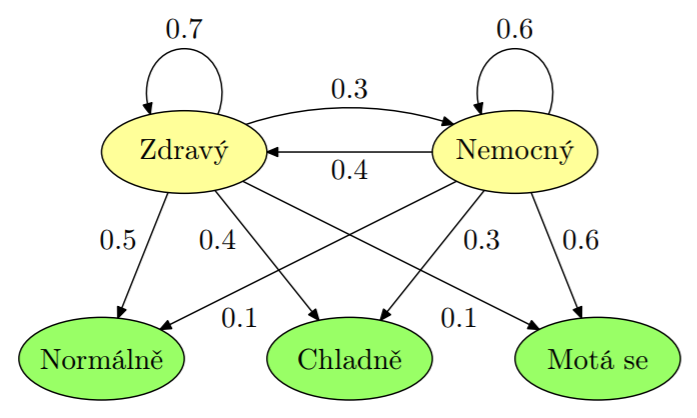
- Známe-li sekvenci pozorování, můžeme spřesnit výpočet pravděpodobnostní distribuce nebo počítat nejpravděpodobnější posloupnost stavů (Viterbiho algoritmus).
- V případě predikce sestřihu si lze zjednodušeně představovat model s dvěma stavy: exon a intron, pro které pozorování jsou určena analyzovanou sekvencí pre-mRNA.
Matematické metody
De Bruijnovy grafy
- Máme slova délky k. Chceme je naskládat přes sebe do řetězce tak, aby dvě po sobě jdoucí slova vždy sdílela k-1 znaků.

- V případě algoritmů pro predikci sestřihu jsou slova úseky ze sekvence, které se algoritmy snaží poskládat dohromady.
- De Bruijnův trik je sestrojit graf, ve kterém jsou slova délky k odpovídají hranám.
- Nalezením Eulerovského tahu poskládáme slova do řetězce.
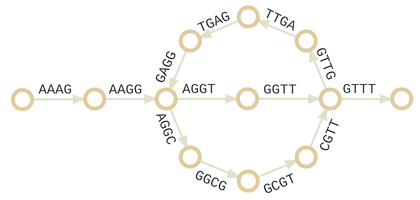
Dvě sekvence: AAAGGTTGAGGCGTTT a AAAGGCGTTGAGGTTT
Dostupné online
Specifická nastavení pro rostliny
AUGUSTUS
Alternativní sestřih
Fgenesh, Fgenesh+, Fgenesh++
GenScan
- Nepředpokládá přesně jeden kompletní gen na vstupu, umožňuje řešit obecné situace, kdy sekvence obsahuje jeden nebo více částečných či kompletních genů.
- Skrytý Markovský model pro hledání kódovaných oblastí. Model bere v úvahu rozdíly v hustotě a struktuře genů. Nevyžaduje dodání podobného genu z proteinové databáze.
- Lze vybrat organismus ze tří možností: obratlovci, Arabidopsis, a kukuřice.
Gnomon
NetGene2
Alternativní sestřih
SOAPdenovo-Trans
Dostupné online
Trinity
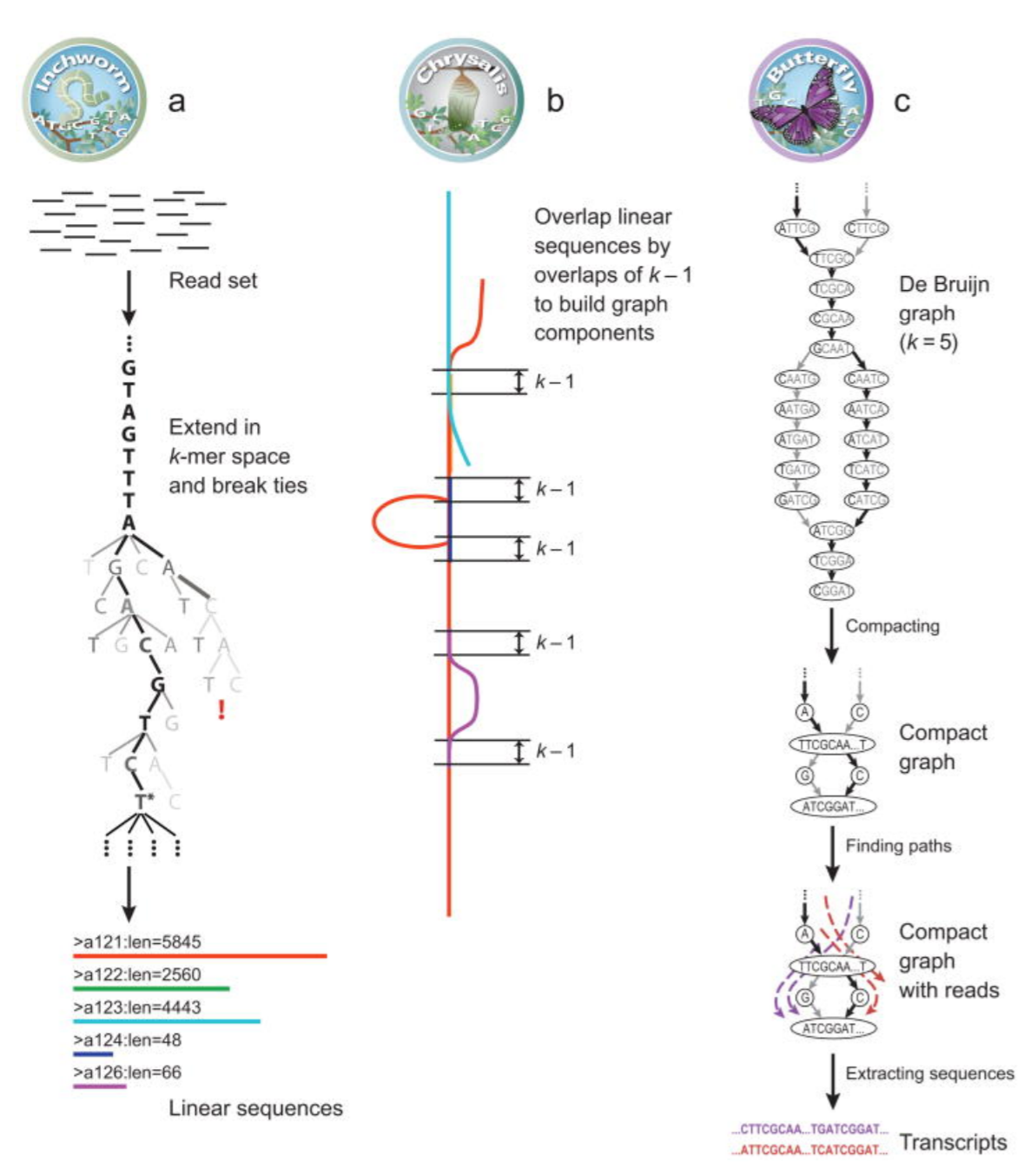
Program je tvořen třemi nezávislými moduly:
- Inchworm rozloží čtení do unikátních kontigů, což jsou sekvence vzniklé zkombinováním čtení.
- Chrystalis shlukuje kontigy do souvislých komponent, pro které sestrojí de Bruijnovy grafy.
- Butterfly analyzuje de Bruijnovy grafy a sestrojuje všechny možné transkripty.
Oases
Cufflinks
- Hledá minimální počet transkriptů, která mohou vysvětlit všechna čtení v grafu.
- Využívá graf překryvů.
PASA
Dostupné online
StringTie
Alternativní sestřih
Lepší český překlad termínu „self-splicing introns“
Otázka: Neexistuje lepší český překlad termínu “self-splicing introns” než „sebe samy vystřihující introny“? Nechť případně autorka navrhne překlad, který nebude zatížen anglicismy.
Použitý překlad je podle článku Stanislava Mihulky ve Vesmíru (https://vesmir.cz/cz/casopis/archiv-casopisu/2001/cislo-2/o-puvodu-eukaryontnich-intronu.html). Není ustálený překlad. Dále se lze se s pojmenováními: introny, které jsou schopny sestřihávat samy sebe, auto-sestřih, auto-splicing.
Asi lepší překlad je „introny vystřihující samy sebe“ nebo „sebevystřihující introny“.
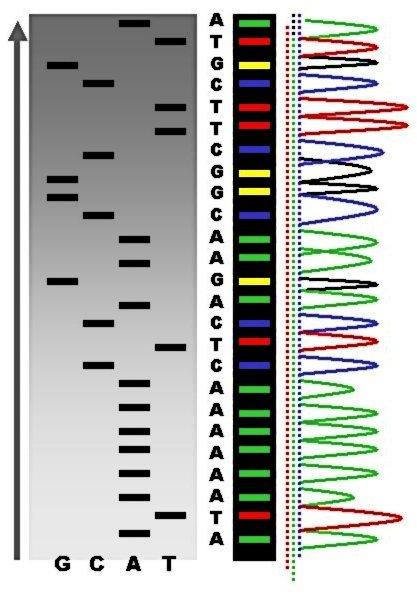
Obrázek 1.7: účel fluorescenčně značených nukleotidů v Sangerově sekvenování
Otázka: Dotaz k obrázku 1.7: K čemu se při uspořádání Sangerova sekvenování s detekcí na gelu, který je popsaný na tomto obrázku, přidávají do reakční směsi fluorescenčně značené nukleotidy?
Abychom mohli detekovat jednotlivé nukleotidy na elektroforéze. Pravdou je, že se toto uspořádání v praxi nepoužívá. Místo toho se využívá kapilární elektroforéza.
Odpovědi na otázky
Výhody sekvenačních metod třetí generace pro získávání transkriptomových dat
Otázka: Autorka zmiňuje potenciál sekvenačních metod třetí generace pro získávání transkriptomických dat. Jaké vidí výhody těchto metod ve srovnání například s paired-end Illumina?
Umožňují rovnou identifikovat exony a introny. Odpádá sestavování transkriptů. Dále by mohly umožnit lepší kvantifikaci transkriptů.
Konkrétní příklady genomických projektů a použitých strategii
Otázka: Práce postrádá konkrétnější informace o použití uvedených algoritmů při sekvenování rostlin, proto by mohla uvést na některých konkrétních genomických projektech, jaká strategie byla použita a zda-li je patrný nějaký trend v používání různých algoritmů.
Sekvenování Suaeda aralocaspica (Wang et al. GigaScience, 8, 2019, 1–9)
Sestavení genomu rýže (Du et al., 2017, Nature Communications)
Referenční genom pro Pisum sativum (Kreplak et al. NATURE GENETICS | VOL 51 | SEPTEMBER 2019 | 1411–1)
- SoapdeNovo2, StringTie, Trinity, PASA (která zahrnuje Evidence Modeler), AUGUSTUS, Fgenesh...
predikce sestřihu
Srovnání algoritmů
- Žádný z algoritmů není univerzálně nejlepší na všechno. Často lze jejich zkombinováním získat lepší výsledky.
- Žádný program pro sestavení transkriptomu nevyužívá kombinaci krátkých a dlouhých čtení. Tyto dva druhy čtení obsahují odlišné informace. Je zajímavým problémem sestrojit algoritmus, který by oba druhy čtení kombinoval.
Přehled algoritmů